





Newsletter Signup - Under Article / In Page
"*" indicates required fields
For advanced therapy medicinal products (ATMPs), the regulatory process is a major challenge to reach the market; but it’s only part of the picture. Here are the top 9 other pre-approval reasons these cell and gene therapies fail.
Sound Scientific Rationale + Good Product = Successful Outcome.” Paula Salmikangas, Committee for Advanced Therapies (CAT) Chairman
1. Narrow Focus
It’s easy to get bogged down in one aspect of development. Some might be tempted to focus on clinical outcomes to attract investment; but in doing so, they might miss or even neglect regulatory issues. Others like Christopher Bravery asks “Do we focus too much on regulatory approval, but not enough on commercialization?”. Frequently, the torpedoes that developers imagine will sink their therapies don’t match those that actually do, as described in this figure:
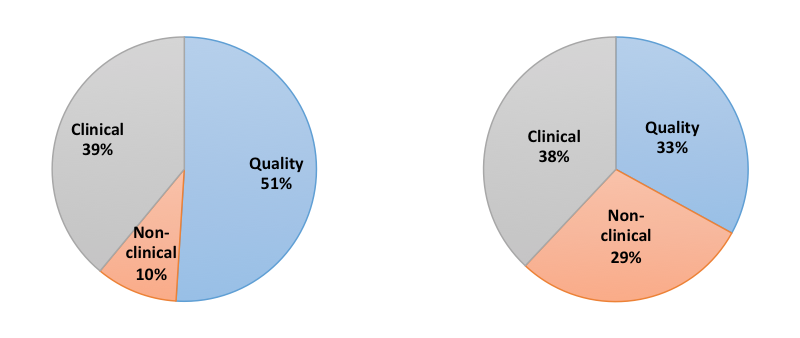
A multidisciplinary approach is therefore critical to product development after the clinical data shows a therapy to be a successful strategy.
2. Product Definition
EMA came up with a dedicated classification procedure to help developers to define their product as ATMPs — that is, cell, gene, and tissue-engineered products. This classification will determine the regulatory strategy, which can be radically different among categories. Last year, Stempeutics’ product was the first stem cell drug based on pooling technology to be classified as ATMP. So far, 244 requests have been submitted since the classification system’s implementation in 2008.
3. Shifting Regulatory Requirements
The lack of regulatory coordination worldwide and within Europe has been cited more than once as a pitfall in ATMP development, as therapies can’t seem to keep up with regulatory developments. When the EU updates its rules, products with years development can be put on hold until they meet the new requirements.
Authorities have taken note, as evidenced by the formation of a joint EMA-FDA committee, but there are still many examples of discord. In 2010, the FDA requested the completion of extra studies on top of EMA requirements before the BLA for Chondrocelect could be filed; further, the FDA asked the developers for a Special Protocol Assessment, which would require additional years of development. Chondrocelect never made it to the US market, and since its market approval in the EU, it has been withdrawn from the market here for commercial reasons.
4. No Risk-Based Approach (RBA)
Historically, many products come from an academic environment without the objective of global commercialization. Consequently, they may not include a strategy to accommodate risk. A risk-based approach, or RBA, is technically optional, but there is no way to make a successful ATMP nowadays without at least an informal one. “Risk-based approach should help developers to overcome challenges due to the specific nature of the ATMPs,” Paula Salmikangas said recently. This planning is particularly important for pioneering treatments that are necessarily “high-risk.” Holoclar, the first stem cell-based product authorized in EU, would not have reached the market if the Chiesi team had not adopted a risk-based strategy.
5. Which Pathway?
It is easy for stakeholders to be lost in the maze of initiatives and tracks to breakthrough. Initiatives like the FDA’s Breakthrough Designation and the EMA equivalent, PRIority MEdicines (PRIME), have been introduced to encourage innovative products addressing unmet medical needs and serious or life-threatening orphan diseases.

Authorities encourage stakeholders to consider these options as early as possible, but there is still a long way to go to streamline these processes. For example, the certification procedure intended to help developers with quality and non-clinical data is a flop: due to the complexity, only 9 treatments have gone this route since its implementation in 2008.
6. Lacking Experience
We can learn from the biologicals industry, which required around 25 years to mature in order to prevent clinical failures. Even if ATMPs market is stammering, there are already sources of data for developers. EMA and CAT release assessment reports (EPAR) for authorized ATMP but thin experience with late-stage development limit any copy/paste strategy. Nonetheless, authorities are open to share gathered experience and tips with developers through a dedicated scientific advice procedure for ATMPs. Since 2009, the CAT has discussed 219 procedures!
7. Quality
Drug quality is normally assessed with the physicochemical paradigm, but it is not applicable for ATMPs, for which new technologies and mindsets are required. There is a much higher number of variables to control in a biological therapy, including sourcing, process variability, cell origin, ability to proliferate, differentiate, generate a response, level of manipulation, manufacturing, route of administration from bench to patient, and potency, which has become particularly important for CAR-T therapies.
For Paula Salmikangas, if a product is poorly standardized and controlled, it may have an impact on product safety and efficacy and lower the chance to get into clinical trials. Holoclar dodged this bullet: its stem cell therapy included non-stem cell phenotypes, but fortunately, Chiesi showed that they do not proliferate.
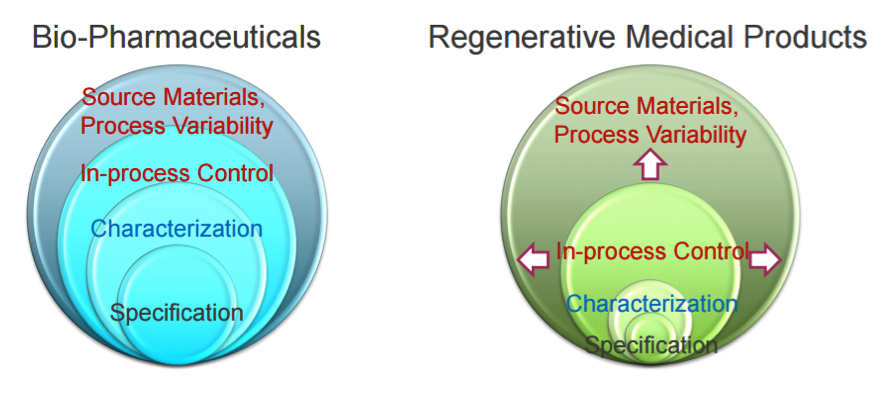
8. Pre-clinical Confusion
Because of the complexity and specificity of cells and tissues, it is much harder to find a suitable model predictive of safety and efficacy for biologicals than for small molecules. In the ATMP framework, tumorigenicity and chromosomal stability are difficult to address during pre-clinical studies.
Therefore, it is key to develop robust methods for tracking cells in vivo in order to address biodistribution, differentiation process and migration in vivo. In this context, large animal models might be necessary sooner than is previously accepted, as was the case for Holoclar. Regenerative medicine is already used in veterinary practice for large animals, and this could be used in future as supportive data.
9. Rushing through the Clinic
“[…] in a period of great confusion about the real therapeutic possibilities of stem cells, such as the one we are living in, being able to demonstrate that stem cells can be definitely safe and successful in a controlled clinical setting is more important than ever,” Chiesi.
In Europe, clinical trials are authorized at national level rather than a continental level, which naturally leads to discrepancies. Promoters may be tempted to go where regulation and costs are reduced, but cutting corners may come back to bite as you get closer to authorization. This pressure to succeed may push developers to rush to Phase III once a positive signal shows up at the Phase II. But if data are not properly gathered there are a high probability that it will go straight into a wall!
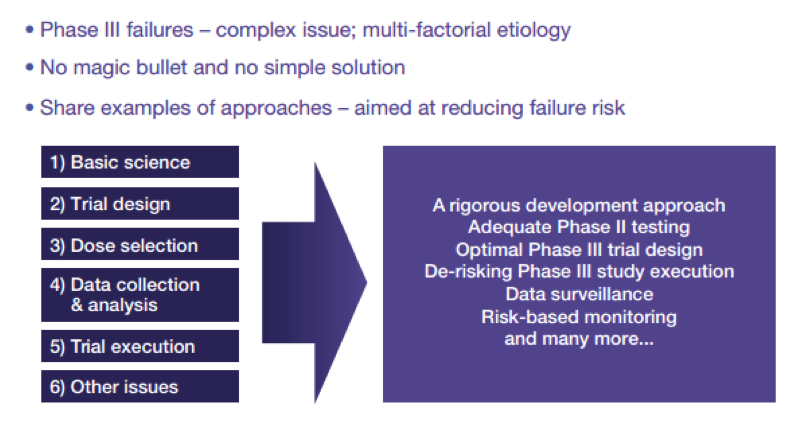
The track from the academic experiment to the market is not set, and the paths of individual therapies may diverge radically. Consequently, risks and expected benefits associated with different products may vary significantly. The challenges relating to the development of ATMPs products are multi-faceted and are therefore difficult to address as a block. At the very least, it can be said that each therapy requires thorough planning and solid research before entering into clinical development. One size does not fit all!
Images from vandame, Freebird7977 / shutterstock.com